What is FSHD?
Facioscapulohumeral muscular dystrophy, or FSHD, is a genetic disorder affecting the skeletal muscles. It typically starts in the early adolescence with the weakness of muscles in the face (facio), shoulders (scapula), upper arms (humerus), and legs. Weakness is slowly progressive and can spread to any muscle. Severity is highly variable among individuals. Approximately 20% of affected individuals eventually require a wheelchair. Life expectancy is not shortened. FSHD is estimated to affect between 4 and 10 individuals per 100,000 people.
Symptoms of FSHD include (but not always):
- Unable to whistle or pucker lips
- Unable to sip through a straw
- Eyes don’t close fully during sleep
- Shoulder blades that “wing” out
- Hard to raise the arms above shoulder
- Difficulty with sit-ups and pull-ups
- Weakness in hands and fingers
- Trouble with rising from a chair
- Difficulty in walking; a waddling gait
- Foot droop
- Protuberant abdomen
- Curved spine
- Chronic fatigue
- Muscle pain
Genetic Cause and Inheritance
FSHD is caused by the abnormal expression of the DUX4 gene, located in the D4Z4 region near the end of chromosome 4 at the 4q35 location. Normally, the DNA in the D4Z4 region includes 11-100 repeated segments of DNA and is hypermethylated, repressing the expression of the DUX4 gene. In FSHD, the expression of DUX4 is reactivated, with two underlying genetic mechanisms.
FSHD1
FSHD2
FSHD2 is clinically identical to FSHD1 but genetically different. In FSHD2, the length of the D4Z4 repeat array on chromosome 4q35 is in normal range. Around 80% of FSHD2 individuals carry a heterozygous mutation in the SMCHD1 gene, and some FSHD2 individuals have mutations in the DNMT3B gene. These mutations are associated with the hypomethylation and relaxation of chromatin in the D4Z4 repeat arrays on both chromosome 4q35 and 10q26. In combination with the 4qA allele, DUX4 is expressed from the D4Z4 repeat array on chromosome 4q35, leading to FSHD. FSHD2 is inherited in a digenic manner.
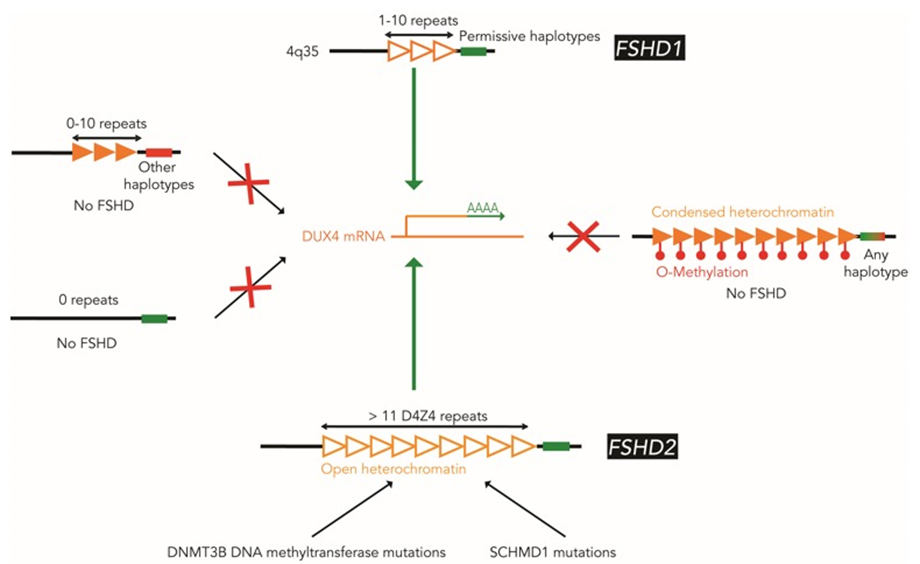
Figure 1: Schematic illustration of FSHD molecular pathogenesis
Diagnosis
FSHD may be diagnosed through a thorough clinical examination, identification of characteristic physical findings, a complete individual and family history, and genetic testing (Figure 2). It usually starts with the testing of FHSD1, which accounts for 95% of the FSHD cases. For many years, the genetic testing of FSHD1 used Southern blot, which is labour and time-intensive, and just provides an estimate of the number of D4Z4 repeats. Pangenia Genomics is the first in Hong Kong to provide the genetic test of FHSD1 based on the Molecular Combing and Genomic Morse Code technology developed by Genomic Vision (http://www.genomicvision.com/products/genetic-tests/fshd/). This method uses specific fluorescent probes to hybridize with genomic DNA molecules (extracted from fresh blood samples) linearly stretched on coverslips (Figure 3). The fluorescent signals can then be visualized to distinguish the length (number of D4Z4 repeat units) and the type (haplotype A or B) of the D4Z4 loci (Figure 4). Most affected individuals have fewer than 10 repeats and the 4qA allele.
Genetic testing for mutations in the SMCHD1 and DNMT3B gene may be indicated if the D4Z4 region on chromosome 4 is not contracted.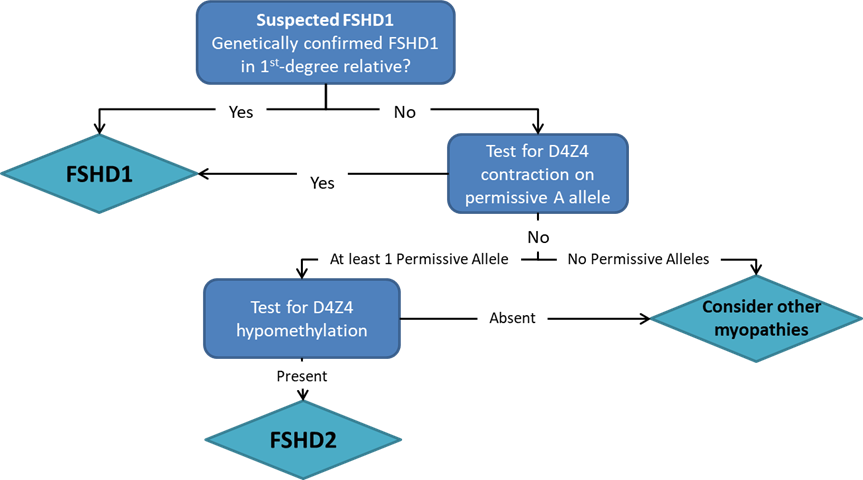
Figure 2: Recommended diagnostic flowchart for FSHD
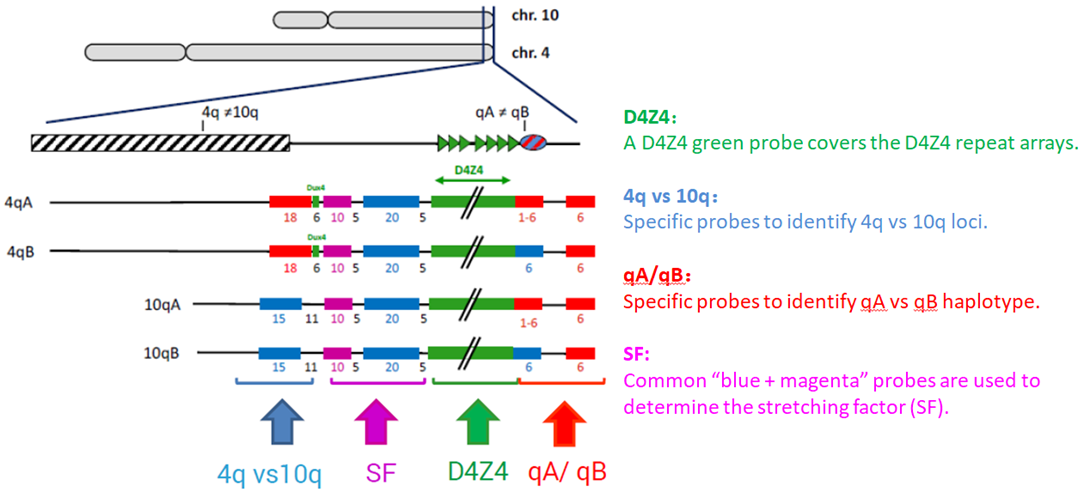
Figure 3: FSHD Genomic Morse Code (GMC) Design
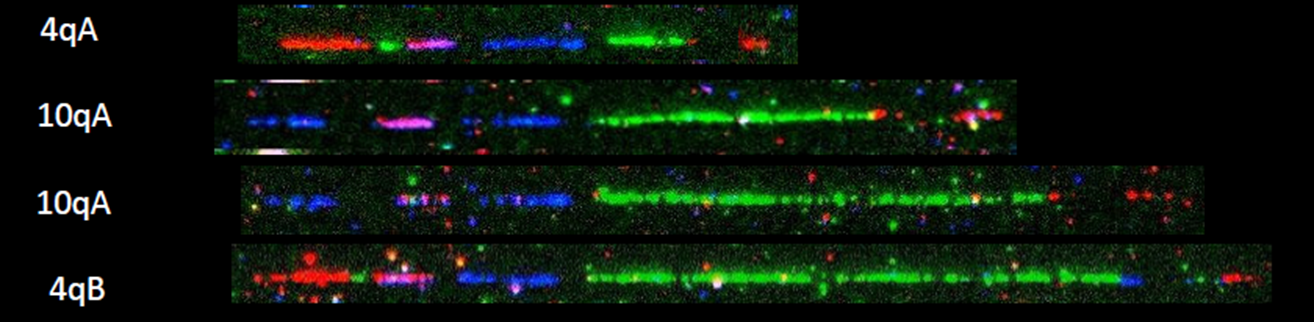
Figure 4: Examples of hybridization signals
Treatments
Currently there is no treatment for FSHD, but several therapies are under investigation. Genetic treatments such as RNAi treatment to silence DUX4 have been evaluated in preclinical studies, though no human trials are currently underway. Losmapimod has been shown in preclinical studies to reduce DUX4 expression, and Phase II trials are currently enrolling for FSHD. Another FSHD treatment candidate GBC0905 is in preclinical studies (https://myocea.com/fshd).
References
https://www.fshdsociety.org/what-is-fshd/
https://www.ncbi.nlm.nih.gov/books/NBK1443/
https://rarediseases.org/rare-diseases/facioscapulohumeral-muscular-dystrophy/