We provide genetic testing services for the following Rare diseases:
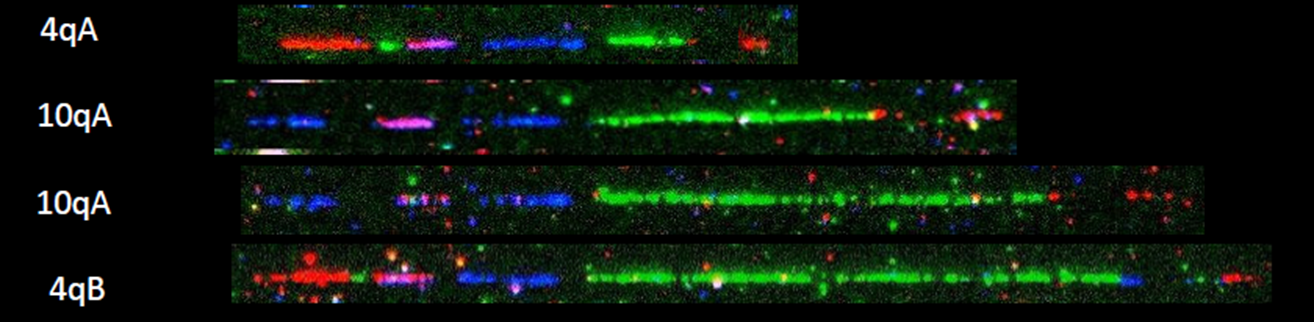
Facioscapulohumeral Muscular Dystrophy (FSHD)
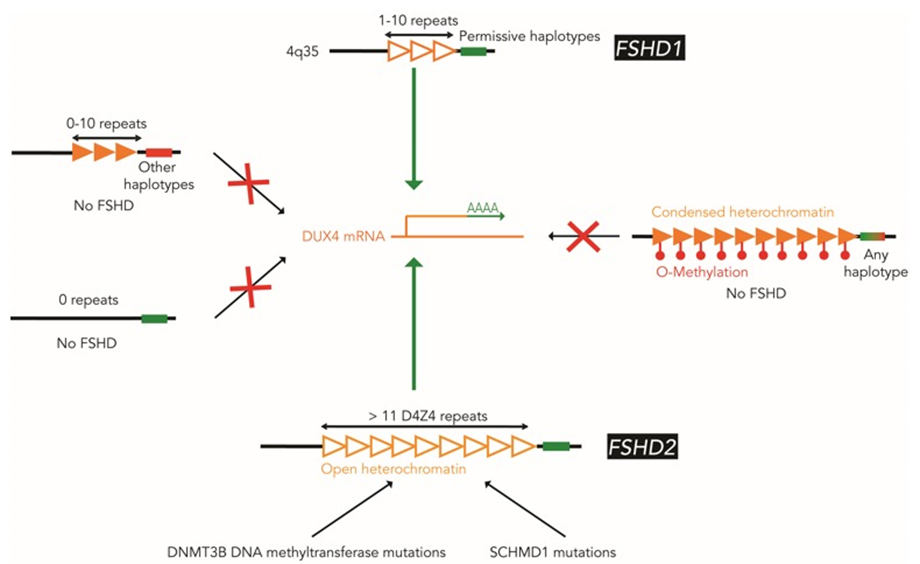
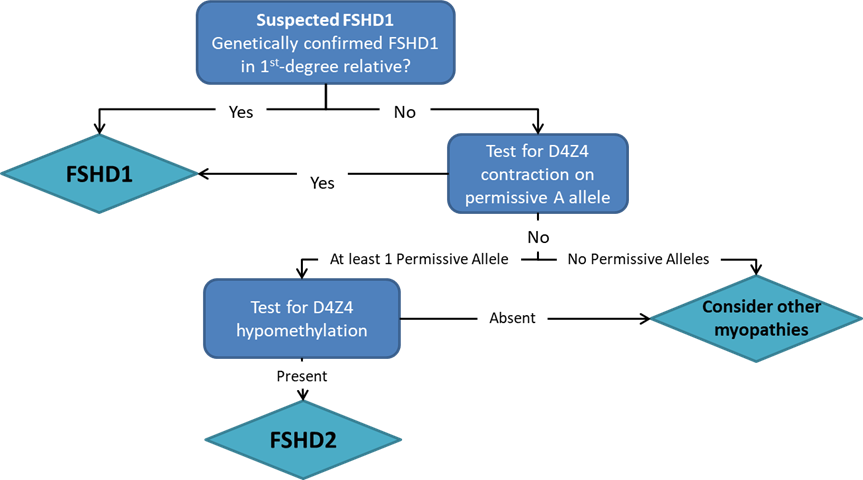
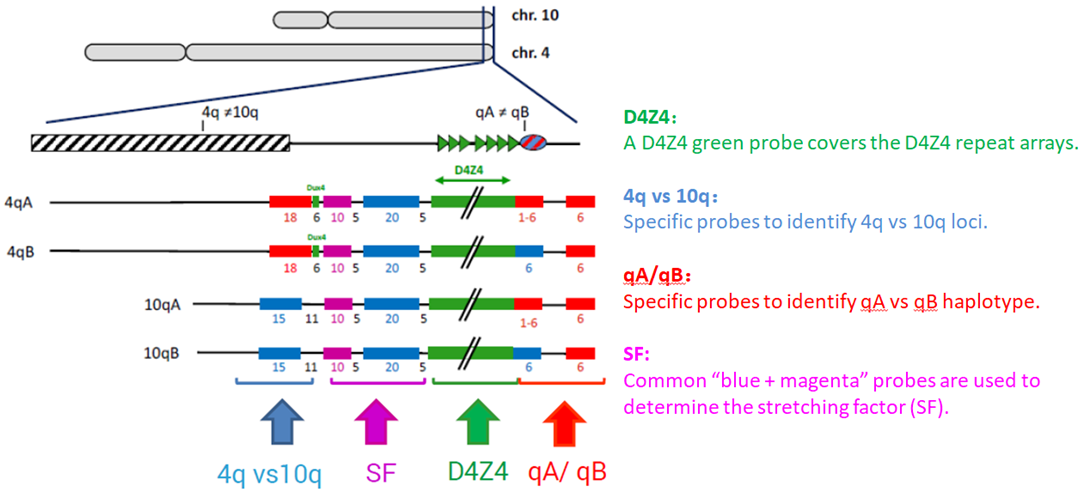
Pangenia Genomics provides the genetic testing of FHSD1 based on the Molecular Combing and Genomic Morse Code technology. This method uses specific fluorescent probes to hybridize with genomic DNA molecules (extracted from fresh blood samples) linearly stretched on coverslips. The fluorescent signals can then be visualized to distinguish the length (number of D4Z4 repeat units) and the type (haplotype A or B) of the D4Z4 loci. Most affected individuals have fewer than 10 repeats and the 4qA allele.
Learn More
Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD)
Pangenia Genomics offers deletion/duplication analysis to accurately detect the number of copies of the DMD gene, i.e. find out if the gene or part of the gene is deleted or duplicated. It can be used for carrier screening as well.
Learn More
Spinal Muscular Atrophy (SMA)
Pangenia Genomics offers deletion/duplication analysis in order to accurately detect deletions and duplications in the SMN1 gene. It can be used for carrier screening as well.
Learn More
Charcot-Marie-Tooth (CMT) disease
Pangenia Genomics offers an in vitro diagnostic (IVD) test for the detection of deletions or duplications in the PMP22, MPZ and GJB1 genes for the diagnosis of Charcot-Marie-Tooth disease type 1 (CMT1) disease. The test can be used to confirm the cause and diagnosis for hereditary neuropathy with liability to pressure palsies (HNPP) as well.
Learn More